 This site is intended for U.S audiences
This site is intended for U.S audiences





This site is intended for U.S audiences
What is Hunter Syndrome (MPS II)?

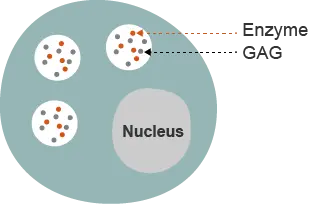
The mucopolysaccharidoses (MPS) are a group of lysosomal storage disorders caused by deficiency of enzymes that catalyze the degradation of glycosaminoglycans (GAGs).1 The MPS disorders are rare diseases and share many clinical features.1 The specific enzyme deficiency present categorizes the MPS disorders into distinct types.1 All types are inherited in an autosomal recessive manner, except MPS II, which is also called Hunter syndrome.1
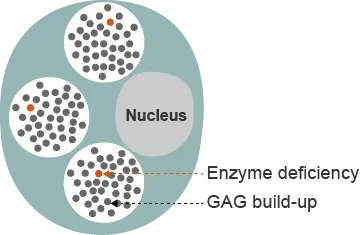
Hunter syndrome is a rare, X-linked, recessive disorder that almost exclusively affects males.1 The estimated prevalence of Hunter syndrome is 1 in 162,000 male live births.2 Hunter syndrome is characterized by a deficiency in the activity of the lysosomal enzyme iduronate-2-sulfatase (I2S), which is one of the lysosomal enzymes responsible for degrading GAGs.3 Undegraded or partially undegraded GAGs progressively accumulate within tissues and organs, causing the multisystemic signs and symptoms of Hunter syndrome.1,4
Hunter syndrome has traditionally been categorized into two forms based on neurological involvement; however, the condition is better regarded as a continuum between the two types: neuropathic and non-neuropathic (previously known as severe and attenuated, respectively).5,6 Two-thirds of patients present with central nervous system involvement, representing the more severe end of the phenotypic spectrum.3
Non-neuropathic
Neuropathic
In patients with the neuropathic type, clinical signs and symptoms typically emerge between 2 and 4 years of age.7 Progressive neurological involvement is prominent and patient mortality may be restricted to the first or second decade of life, usually due to obstructive airway disease or cardiac failure.8
At the opposite end of the spectrum, patients with the non-neuropathic type may not present with symptoms until late childhood or early adolescence, and neurologic dysfunction is minimal.7,8 These patients may survive into adulthood.8
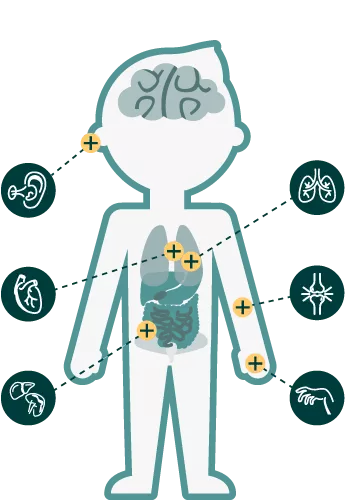
Early Neurological Signs/Symptoms in MPS Patients
1. Neufeld EF, Muenzer J. In: Scriver CR, eds. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill; 2001:3421-3452. 2. Meikle PJ et al. JAMA. 1999;281(3):249-254. 3. Wraith JE et al. Eur J Pediatr. 2008;167(3):267-277. 4. Muenzer J et al. Pediatrics. 2009;124(6):e1228-e1239. 5. Giugliani R et al. Genet Mol Biol. 2014;37(2):315-329. 6. Scarpa M et al. Orphanet J Rare Dis. 2011;6:72. 7. Burton BK, Giugliani R. Eur J Pediatr. 2012;171(4):631-639. 8. Martin R et al. Pediatrics. 2008;121(2):e377-e386.
Hunter syndrome is a
progressive genetic disease
If you suspect Hunter syndrome, refer your patient to a metabolic geneticist for an accurate diagnosis.